
Pharmacokinets: Drug metabolism or Drug Biotransformation
Introduction to Biotransformation or Drug Metabolism
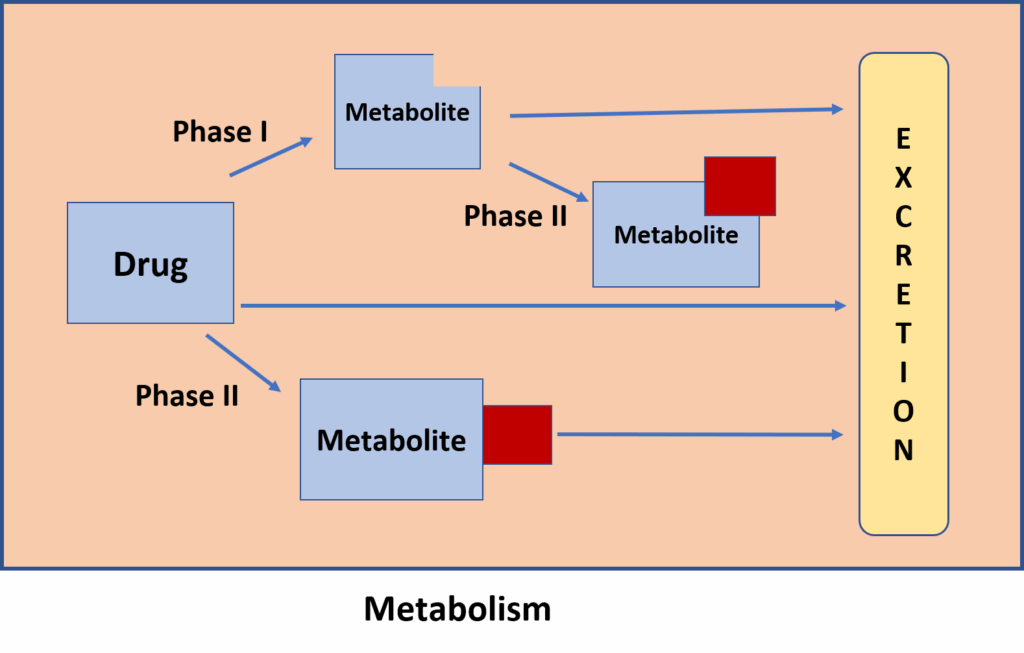
Drug metabolism, or Biotransformation, is the biochemical process through which the body chemically alters drugs. This transformation typically occurs to convert lipophilic (fat-soluble) substances into more hydrophilic (water-soluble) compounds. The reason for this transformation is that lipophilic substances, if not altered, tend to be reabsorbed in the renal tubules and not excreted effectively. Hydrophilic compounds, on the other hand, are readily excreted through urine. Therefore, biotransformation is crucial for drug elimination. This mechanism has evolved in humans to defend against naturally ingested toxins. While many drugs are extensively metabolised, others like streptomycin and neostigmine undergo minimal transformation and are excreted unchanged.
Sites of Biotransformation in the Body
The liver is the primary site for drug metabolism due to its rich supply of drug-metabolising enzymes. However, other organs like the intestines, kidneys, lungs, and plasma also contribute. These organs collectively ensure that drugs are metabolised and eliminated efficiently. This biotransformation may result in different outcomes:
Inactivation: Most drugs and their metabolites become less active or inactive after metabolism (e.g., ibuprofen, lidocaine).
Active Metabolites from Active Drugs: Some drugs produce additional active metabolites, which contribute to the overall therapeutic effect.
Prodrugs: Certain drugs are administered in an inactive form and require metabolic conversion to become active (e.g., enalapril to enalaprilat). Prodrugs can be advantageous due to improved absorption, reduced side effects, or targeted activation.
Phases of Drug Metabolism
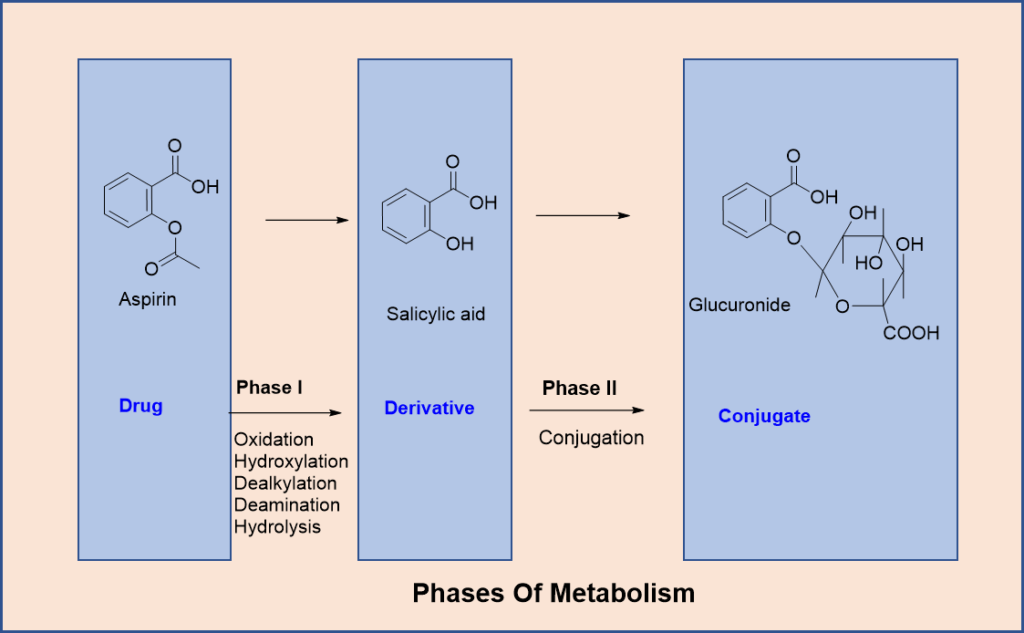
Drug metabolism occurs in two distinct phases:
1. Phase I (Functionalization Reactions): These involve chemical modifications such as oxidation, reduction, or hydrolysis. The result is often a molecule with an exposed or added functional group that prepares it for Phase II reactions.
2. Phase II (Conjugation Reactions): In this phase, an endogenous molecule like glucuronic acid or sulfate is added to the drug or its Phase I metabolite. This increases its water solubility and facilitates excretion. These reactions generally lead to inactive metabolites, though there are exceptions.
Phase I Reactions
Oxidation is the most frequent Phase I reaction. It includes reactions like hydroxylation, dealkylation, deamination, and others. These are primarily carried out by cytochrome P-450 (CYP450) enzymes, found in the liver’s smooth endoplasmic reticulum. There are over 100 CYP450 isoenzymes, with CYP3A4, CYP2D6, and CYP2C9 being among the most significant for drug metabolism.
Reduction reactions are the reverse of oxidation and often occur under low oxygen conditions. Drugs such as warfarin and halothane undergo reduction.
Hydrolysis involves breaking bonds in esters and amides using water, catalysed by enzymes like esterases and amidases. Drugs such as aspirin and lidocaine are metabolised this way.
Cyclisation and decyclisation are less common reactions that involve the formation or opening of ring structures in drug molecules.
Phase II Reactions
Glucuronidation is the most prevalent conjugation reaction, involving UDP-glucuronosyl transferase (UGT). Drugs like morphine and paracetamol are metabolised through this pathway.
Acetylation adds an acetyl group using acetyl-CoA and is especially important for drugs with amino groups, like isoniazid and sulfonamides. Genetic polymorphism in acetylation capacity leads to classification as slow or fast acetylators.
Methylation, Sulfation, and Glycine Conjugation are additional Phase II reactions that modify amines, phenols, and carboxylic acids, respectively.
Glutathione Conjugation is crucial for detoxifying reactive metabolites like those formed from paracetamol overdose.
Ribonucleotide Synthesis is used in the activation of anticancer agents like purine and pyrimidine analogues.
Variability in Drug Metabolism
Drug metabolism is influenced by both genetic and environmental factors. For example, individuals may differ in CYP2D6 activity, making them poor or extensive metabolizers. Stereoisomers like S-warfarin and R-warfarin also exhibit differences in metabolism. Interspecies differences are significant too—cats lack UGT enzymes, whereas dogs are deficient in NATs. This variability contributes to differences in drug efficacy and safety.
Types of Drug-Metabolising Enzymes
Microsomal Enzymes (e.g., cytochrome P450s, UGTs) are found in the liver’s smooth endoplasmic reticulum and are responsible for most oxidation, reduction, and conjugation reactions. They are inducible by drugs, diet, and environmental factors.
Nonmicrosomal Enzymes are located in the cytoplasm and mitochondria of liver cells and are responsible for many hydrolytic and conjugation reactions, excluding glucuronidation. These enzymes are not typically inducible but can exhibit genetic polymorphism.
Biotransformation in Neonates
Newborns, particularly preterm infants, have underdeveloped enzyme systems. This makes them more susceptible to drug toxicity, such as gray baby syndrome from chloramphenicol. While Phase I reactions mature rapidly within months, Phase II reactions like glucuronidation may take longer to develop.
Enzyme Induction and Its Effects
Certain substances can increase the expression of metabolic enzymes. For instance:
(i) Phenobarbital and rifampin induce CYP3A4, accelerating the metabolism of many drugs.
(ii) Chronic alcohol use induces CYP2E1, enhancing the formation of toxic metabolites from drugs like paracetamol.
Consequences include reduced drug efficacy (e.g., oral contraceptive failure), increased toxicity, development of tolerance, and altered metabolism of endogenous substances like steroids.
Clinical Applications and Concerns
Enzyme induction has therapeutic and toxicological implications. For example:
(i) Phenobarbital can treat neonatal jaundice by enhancing bilirubin metabolism.
(ii) Rifampin-induced metabolism may require dose adjustments in co-administered drugs.
(iii) Drug interactions may arise when one drug induces enzymes that metabolise another.
First Pass Metabolism
First-pass metabolism refers to the breakdown of orally administered drugs in the gut wall and liver before reaching systemic circulation. Drugs with high first-pass metabolism (e.g., propranolol, nitroglycerin) have reduced bioavailability. This can be avoided by using alternative routes like sublingual or intravenous administration. Understanding this helps in proper dosing and drug formulation.
References
Latest Editions of
- Rang H. P., Dale M. M., Ritter J. M., Flower R. J., Rang and Dale’s Pharmacology,.Churchil Livingstone Elsevier
- K.D.Tripathi. Essentials of Medical Pharmacology, JAYPEE Brothers Medical Publishers (P) Ltd, New Delhi.
- Sharma H. L., Sharma K. K., Principles of Pharmacology, Paras medical publisher